
Every time you pick up a prescription and see a lower price tag than the brand-name version, you’re seeing the ANDA process at work. The Abbreviated New Drug Application isn’t just bureaucratic jargon-it’s the reason 9 out of 10 prescriptions filled in the U.S. are generic drugs. And those generics aren’t cheap knockoffs. They’re exact copies in active ingredients, strength, and how they work in your body-approved by the FDA without repeating every single clinical trial ever done for the brand-name drug.
What Exactly Is an ANDA?
The ANDA, or Abbreviated New Drug Application, is the official pathway generic drug makers use to get FDA approval. It’s called ‘abbreviated’ because it doesn’t require the applicant to prove the drug is safe and effective from scratch. Instead, they only need to show their version is bioequivalent to the original brand-name drug, known as the Reference Listed Drug (RLD). That means the generic delivers the same amount of active ingredient into your bloodstream at the same speed as the brand. No extra studies on animals or large human trials needed.
This system was created by the Hatch-Waxman Act in 1984. Before that, generic companies had to start from zero-spending years and hundreds of millions just to prove a drug worked. Hatch-Waxman changed everything. It gave generics a clear, faster route while still protecting the original innovator’s patents. The result? Generic drugs now save the U.S. healthcare system over $370 billion every year.
How the ANDA Process Works Step by Step
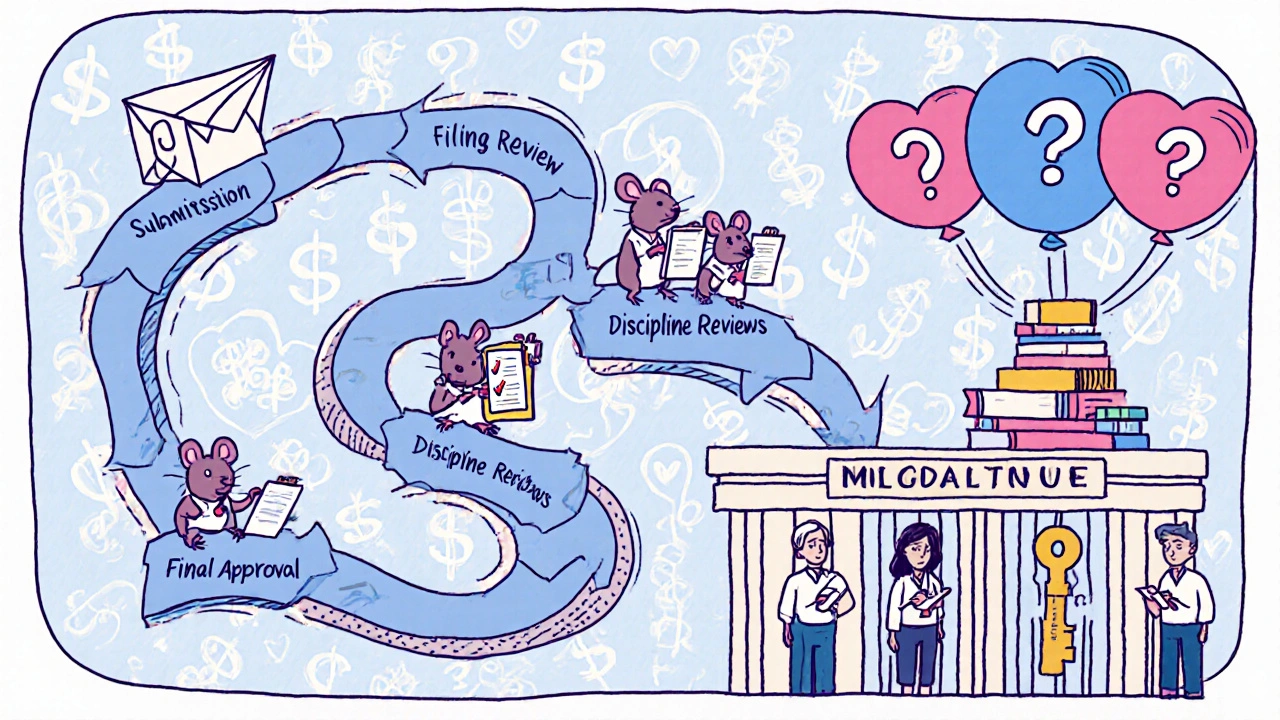
The FDA’s Office of Generic Drugs (OGD) handles all ANDA submissions. Here’s how it actually plays out:
- Submission: The company files an electronic application using FDA’s Electronic Submission Gateway. It includes detailed chemistry data, manufacturing details, labeling, and bioequivalence study results. The form FDA-356h is mandatory.
- Filing Review: Within 60 days, the FDA checks if the application is complete enough to even be reviewed. If it’s missing key pieces-like a proper bioequivalence study-it gets sent back.
- Discipline Reviews: Teams of scientists dig into every part: chemistry, manufacturing, microbiology, labeling, and bioequivalence. Each team looks for compliance with FDA standards. This is where most applications get stuck.
- Information Requests: If something’s unclear or missing, the FDA sends an Information Request (IR). Most applicants get between 10 and 20 of these. Answering them correctly takes time, often months.
- Final Decision: If everything checks out, you get Final Approval. But if there’s a patent or exclusivity blocking the drug from hitting the market yet, you get Tentative Approval. You’re cleared scientifically, but you can’t sell it until the patent expires.
Under GDUFA III (the current user fee agreement), the FDA aims to finish its first review of an original ANDA in 10 months. But the whole process-from filing to market-often takes 30 months because of back-and-forth requests and inspections.
What Makes a Generic ‘Bioequivalent’?
It’s not enough to say, ‘It has the same pill inside.’ The FDA requires hard data. For most oral tablets, that means a bioequivalence study with 24 to 36 healthy volunteers. They take the brand and the generic on different days, and blood samples are taken over 24 to 72 hours. The FDA looks at two key numbers: how much of the drug gets into the blood (AUC) and how fast it gets there (Cmax).
The generic’s results must fall within 80% to 125% of the brand’s. That’s the sweet spot. If it’s outside that range, the FDA says it’s not equivalent. For complex products like inhalers, creams, or injectables, this gets harder. The body doesn’t absorb them the same way every time. That’s why 35% of Complete Response Letters from the FDA in 2022 cited problems with bioequivalence studies.
One company spent $1.2 million and three tries to get their topical cream approved. That’s not unusual. Complex generics are the fastest-growing part of the ANDA pipeline now-making up 35% of all pending applications.
ANDA vs. NDA: Why Generic Is So Much Cheaper
Here’s the real contrast:
| Feature | ANDA (Generic) | NDA (Brand) |
|---|---|---|
| Required Data | Bioequivalence only; relies on FDA’s prior findings for RLD | Full preclinical and clinical trials from scratch |
| Typical Cost | $1 million to $5 million | $2.3 billion |
| Development Time | 3-5 years (including preparation) | 10-15 years |
| Patent Protection | No; must navigate existing patents | Up to 20 years |
| Approval Rate (First Cycle) | 91% | ~30% |
The numbers don’t lie. A brand-name drug company spends billions and over a decade to get one drug approved. A generic company spends a fraction of that and gets a similar product to market in a fraction of the time. That’s why generic drugs cost, on average, only 15% of the brand’s price once they hit the market.
Common Pitfalls and Why Applications Get Rejected
Even with a clear roadmap, many ANDAs fail. The top three reasons:
- Bioequivalence issues (35% of rejections): Poor study design, small sample sizes, or not following FDA’s product-specific guidance.
- Manufacturing problems (28%): Facilities not compliant with cGMP (current Good Manufacturing Practices). The FDA inspects every plant listed in the ANDA. If one factory fails, the whole application stalls.
- Labeling errors (22%): The generic’s label must match the brand’s exactly-except for the name and manufacturer. Missing warnings, wrong dosage instructions, or even font size issues can trigger a rejection.
Companies that succeed early often use pre-ANDA meetings with the FDA. These are optional, but 78% of top applicants use them. It’s like asking the referee for clarification before the game starts. You avoid costly mistakes.
Who’s Playing in This Game?
The generic drug market is dominated by a few big players: Teva Pharmaceuticals (22% market share), Viatris (15%), and Sandoz (12%). But 75% of all ANDAs come from companies that have already approved five or more generics. That’s because experience matters. The first time you file, you’re learning the system. By the fifth or tenth, you’ve built internal teams, templates, and processes that cut review time.
One VP at Teva said after their tenth ANDA, they hit GDUFA timelines 92% of the time. That’s not luck-it’s process. They know what the FDA’s looking for before they even submit.
The Future of Generic Drugs
The FDA is adapting. With more complex generics-like inhalers, nasal sprays, and injectables-on the rise, they’re rolling out over 450 new product-specific guidances every year. They’re also using AI to help review chemistry data faster. Over 78% of FDA reviewers now use AI-assisted tools to spot inconsistencies in data.
But challenges remain. Patent thickets-where brand companies pile on multiple patents to delay generics-are still a major barrier. So are REMS programs (Risk Evaluation and Mitigation Strategies), which can restrict access to brand drugs for testing. Without the brand drug, you can’t run the bioequivalence study.
Still, the numbers are clear: the ANDA process works. It’s not perfect, but it’s the engine behind America’s affordable medicine supply. Without it, millions of people couldn’t afford their prescriptions. And that’s why it’s not just a regulatory process-it’s a public health tool.
What You Need to Know as a Patient
If your doctor prescribes a brand-name drug, ask if there’s a generic. It’s not just cheaper-it’s the same medicine. The FDA requires generics to meet the same quality standards as brands. The only differences are the color, shape, or inactive ingredients like fillers-none of which affect how the drug works.
Some people worry generics aren’t as strong. That’s a myth. The FDA doesn’t approve a generic unless it delivers the same amount of active ingredient into your blood as the brand. If you’ve ever switched from a brand to a generic and felt a difference, it’s likely psychological-or maybe you’re sensitive to a filler. But the active ingredient? It’s identical.
And if you’re ever unsure? Check the FDA’s Orange Book. It lists all approved generics and their RLDs. You’ll find the exact match.
Is a generic drug the same as the brand name?
Yes, legally and scientifically. A generic drug must have the same active ingredient, strength, dosage form, route of administration, and therapeutic effect as the brand-name version. The FDA requires bioequivalence testing to prove this. The only differences are in inactive ingredients, packaging, or brand name-none of which affect how the drug works in your body.
Why do generic drugs cost so much less?
Generic manufacturers don’t have to repeat expensive clinical trials. They rely on the FDA’s prior approval of the brand-name drug’s safety and effectiveness. This cuts development costs from over $2 billion to around $1-5 million. Lower costs mean lower prices-and competition among multiple generic makers drives prices down even further, often to 15% of the brand’s price.
How long does the ANDA process take?
The FDA aims to complete its first review within 10 months under GDUFA III. But the full process-from preparing the application to final approval-typically takes 2.5 to 3 years. Delays often come from Information Requests, manufacturing inspections, or patent disputes. Tentative Approval can happen faster if the only barrier is a patent that hasn’t expired yet.
What’s the difference between Tentative Approval and Final Approval?
Tentative Approval means the FDA has found the generic meets all scientific and manufacturing requirements-but it can’t be sold yet because of an active patent or exclusivity period on the brand-name drug. Final Approval means all scientific, legal, and manufacturing hurdles are cleared, and the drug can be marketed immediately.
Are all generic drugs made in the U.S.?
No. Over 80% of generic drug ingredients are manufactured overseas, mostly in India and China. But every facility-whether in the U.S., India, or elsewhere-must pass an FDA inspection before approval. The FDA inspects over 3,500 foreign facilities each year. The drug’s origin doesn’t affect its quality if it’s FDA-approved.
Can a generic drug be recalled?
Yes. Generic drugs are subject to the same recall rules as brand-name drugs. If a quality issue is found-like contamination, incorrect labeling, or manufacturing defects-the FDA can order a recall. In fact, some of the largest recalls in recent years have been for generic drugs, often tied to facility violations or impurities in the active ingredient.
What’s Next for Generic Drugs?
The future of generics isn’t about more pills-it’s about smarter ones. The FDA is pushing for better ways to approve complex generics: inhalers that deliver medicine deep into the lungs, injectables with nanoparticles, and topical creams that penetrate skin layers consistently. These aren’t easy to copy. But with new science and AI tools, the system is adapting.
One thing won’t change: the ANDA process remains the backbone of affordable medicine in the U.S. It’s not flashy. It doesn’t make headlines. But every time someone fills a prescription and pays less, it’s because of this quiet, science-driven system that’s been saving lives-and money-for over 40 years.




Manjistha Roy
November 22, 2025 AT 23:49The ANDA process is one of those quiet miracles of modern medicine-no one notices it until they can’t afford their pills. The fact that a generic version of a $500 drug can cost $75 isn’t magic; it’s the result of decades of careful regulatory design. I’ve worked in pharma compliance in India, and I can tell you: the quality control standards the FDA enforces on foreign manufacturers are brutal, but necessary. Many Indian plants spend more on compliance than on R&D. Still, it works. Millions of diabetics in rural India take FDA-approved metformin every day. That’s not a coincidence-it’s policy.
JD Mette
November 24, 2025 AT 09:01I’ve switched to generics for years and never had an issue. My cardiologist told me once that the only difference between brand and generic lisinopril is the filler-and sometimes, the brand’s filler gives me heartburn. So I stick with generic. Simple as that. The FDA doesn’t mess around with bioequivalence. If it passes, it’s safe.
Olanrewaju Jeph
November 25, 2025 AT 13:41It is important to recognize that the Hatch-Waxman Act was a landmark piece of legislation that successfully balanced innovation with accessibility. Without it, the pharmaceutical industry would have remained monopolistic, and patients would have been forced to pay exorbitant prices for life-saving medications. The ANDA process is not merely a regulatory shortcut-it is a strategic mechanism that promotes competition, reduces healthcare expenditures, and ultimately saves lives. The data supporting this are unequivocal.
Dalton Adams
November 25, 2025 AT 19:57Look, most people don’t realize that 80% of generic APIs come from China and India, and the FDA inspects maybe 10% of those facilities annually. That’s not oversight-that’s a gamble. I’ve seen lab reports where the active ingredient varied by 18% between batches. The FDA’s 80–125% bioequivalence window? That’s a legal loophole disguised as science. If your drug’s absorption varies by 25%, it’s not the same-it’s a different pharmacokinetic profile. And don’t get me started on the impurities in metformin from certain manufacturers. Some batches had NDMA levels above WHO limits. The FDA didn’t recall them all. They just updated the guidance. Classic.
Kane Ren
November 26, 2025 AT 08:17This is why I believe in the system. Even with all the bureaucracy, it still delivers. I used to pay $400 a month for my asthma inhaler. Now? $12. Same medicine. Same relief. No magic, no conspiracy-just smart regulation. The FDA isn’t perfect, but they’re trying. And honestly? That’s more than most industries can say.
Charmaine Barcelon
November 27, 2025 AT 07:21People are so naive. They think generics are just as good. But have you ever switched and felt weird? Dizzy? Nauseous? That’s not placebo. That’s your body reacting to different fillers, different binders, different coatings. The FDA says it’s fine-but they don’t test how you feel. They test blood levels. That’s not medicine. That’s math. And math doesn’t care if you can’t sleep at night.
Karla Morales
November 28, 2025 AT 14:42📊 The data is clear: 91% of ANDAs pass first review. But here’s what they don’t tell you: 78% of those approved are from just 5 companies. Teva, Viatris, Sandoz, Dr. Reddy’s, and Sun Pharma. The rest? They’re the noise. The FDA’s AI tools are trained on data from these giants. So when a small manufacturer submits? The system is biased toward scale. It’s not about quality-it’s about who can afford to play the game. Also, 🤔 why are 35% of rejections tied to bioequivalence? Because the testing protocols are outdated. We’re still using 1980s blood sampling for 2020s nanotech drugs. 🚨
Javier Rain
November 29, 2025 AT 15:12Let’s be real-this whole system is a win for patients. I’ve seen friends choose between insulin and groceries. Generics changed that. Yes, there are hiccups. Yes, some plants cut corners. But the FDA’s inspections are getting smarter, faster, and more transparent. And the fact that we’re using AI to catch data inconsistencies? That’s progress. We’re not perfect, but we’re moving forward. Keep pushing for better oversight, but don’t throw out the system that’s already saving billions.
Laurie Sala
November 30, 2025 AT 01:43I switched to a generic antidepressant last year... and I felt like a different person. Like, emotionally numb. Not depressed-just... empty. I went back to the brand, and suddenly, I was crying again-happy tears, even. I know the FDA says it’s the same... but my body knew the difference. I don’t care about the science. I care about feeling alive. And that generic stole that from me.
Lisa Detanna
December 1, 2025 AT 07:10I’m from Nigeria, and I’ve seen how expensive medicines are there. When I came to the U.S., I was shocked that my blood pressure meds cost $4. I didn’t know how it worked-until I read this. The ANDA system isn’t just American-it’s a global model. India and China export billions in generics because of this framework. It’s not perfect, but it’s the most equitable system we’ve ever had for medicine. We should be protecting it, not tearing it down.
Demi-Louise Brown
December 3, 2025 AT 02:10Generic drugs are not a compromise. They are a necessity. The cost savings allow public health programs to cover more patients. They allow elderly Americans on fixed incomes to stay on their medications. They allow parents to choose treatment over bankruptcy. The science is solid. The data is overwhelming. The FDA’s standards are rigorous. The system works. Let’s not let fear or misinformation erode what has become a cornerstone of accessible healthcare.
Matthew Mahar
December 4, 2025 AT 04:44so like... i just found out that my generic xanax is made in a factory in hyderabad and shipped through a warehouse in new jersey?? like... i just assumed it was made in the usa?? and now i dont know if i should be scared or impressed?? 🤯
John Mackaill
December 4, 2025 AT 05:05There’s a reason the U.S. is the largest consumer of generics in the world. It’s not because we’re cheap-it’s because we’re rational. The ANDA process allows competition to drive prices down without sacrificing safety. Other countries don’t have this balance. The UK’s NHS pays more per pill than we do for the same drug. Why? Because they don’t have a functional generic pathway. We should be proud of this system-not cynical about it.
Adrian Rios
December 4, 2025 AT 16:49Let me tell you something most people don’t get: the ANDA process is the only reason we have affordable insulin. Without it, the three big brand makers-Eli Lilly, Novo Nordisk, Sanofi-would still be charging $300 a vial. The first generic insulin was approved in 2020. It cost $25. And guess what? The brand companies didn’t collapse. They lowered their prices. That’s capitalism working the way it should. Competition doesn’t kill innovation-it forces it to be more responsible. And the FDA? They didn’t just sit there. They rewrote the bioequivalence guidelines for biologics. That’s leadership. That’s what happens when science meets policy. This isn’t bureaucracy-it’s justice.
Casper van Hoof
December 6, 2025 AT 02:45One might posit that the ANDA framework constitutes a form of epistemological economy: the state, by virtue of its prior validation of the innovator’s drug, externalizes the burden of proof onto the generic manufacturer, thereby reducing transactional costs while preserving epistemic integrity. The bioequivalence standard, though statistically derived, functions as a hermeneutic bridge between proprietary knowledge and public access. Thus, the FDA does not merely regulate-it mediates between the metaphysics of pharmaceutical innovation and the material exigencies of public health. In this light, the ANDA is less a procedure than a philosophical compromise-a social contract written in pharmacokinetic curves.